Kairos derep-detect
see Installation for installing
Description
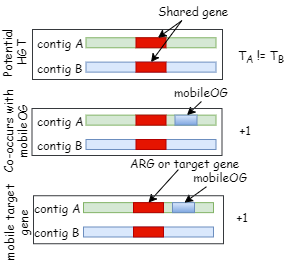
Kairos assess detects identical genes in distinct genetic contexts. Kairos derep-detect identifies identical orfs in contigs and assesses their redundancy based on the proportion of orfs that are shared to those that are unique. Putative HGTs are those events where identical orfs occur in two contigs with differing taxonomic assignment. Potential HGTs are scored based on whether the putatively transfered gene is a target gene and/or co-localized with an MGE hallmark gene.
Quick Start
To run derep-detect:
(.venv) $ nextflow kairos-dd.nf --max_cpus 128 --max_overlap 0.5 --input_contigs input.fasta --taxa_df taxadf.tsv --outdir output --target_database ${database_dir_path}/deeparg.fasta --MGE_database ${database_dir_path}/mobileOG-db_beatrix-1.6.All.faa --num_chunks 128
Input
inputs to Kairos derep-detect are a fasta file of contigs, taxonomic assignments of contigs, a target gene database (by default, deepARG-db is recommended) and a database of MGE hallmark genes (mobileOG-db is recommended).
input files and required parameters:
taxa_df = null
input taxonomy dataframe of format: contig classification
input_contigs = null
input contigs in fasta format
outprefix = ‘kairos’
job title that will be used for file naming
clustering and detection parameters
mmseqs_prot_cov = 0.3
the mmseqs -c parameter used during identical orf identification
mmseqs_prot_id = 0.99
the mmseqs –id parameter used during identical orf identification
mmseqs_prot_cov_mode = 1
the mmseqs –cov-mode parameter used during identical orf identification
mmseqs_contig_cov = 0.6
the mmseqs -c parameter used during initial contig dereplication
mmseqs_contig_id = 0.99
the mmseqs -c parameter used during initial contig dereplication
mmseqs_contig_cov_mode = 1
the mmseqs -c parameter used during initial contig dereplication
max_overlap = 0.5
the maximum proportion of shared ORFs between two contigs to be considered non-redundant
min_orfs = 1
minimum number of orfs in a contig to consider for HGT analysis
database input commands
target_database=null
absolute path to target database (deepARG-db by default)
MGE_database=null
absolute path to MGE database (mobileOG-db by default)
diamond alignment parameters
MGE_id = 0.3
identity value for MGE annotation
MGE_e = 1e-5
e-value for MGE annotation
target_id = 80
identity value for target annotation
target_e = 1e-10
e-value for target annotation
target_query_cover = 0.8
query-cover parameter for target annotation
max_dist_closest_MGE = 5000
the closest MGE must be within this basepair distance in order to score +1 on MGE colocalization
Output
Table 1. Output files and descriptions for Kairos derep-detect.
Output File |
Description |
*_target_dmnd.tsv |
Diamond table of target matches |
*_MGE_dmnd.tsv |
Diamond table of MGE matches |
phylum_HGT.csv |
Phylum-level HGTs |
class_HGT.csv |
Class-level HGTs |
order_HGT.csv |
Order-level HGTs |
family_HGT.csv |
Family-level HGTs |
genus_HGT.csv |
Genus-level HGTs |
species_HGT.csv |
Species-level HGTs |
kairos_deduplicated_overlaps.tsv |
Contigs with nonredundant contigs |
kairos_overlap_out.tsv |
Merged overlapping contigs output file |
kairos_redundant_overlaps.tsv |
Redundant contigs |
kairos_contig_clusters.tsv |
Contig cluster assignments |
kairos_overlap_log.txt |
Log file for overlap detection |
kairos_clustering_log.txt |
Log file for clustering steps |
Extended Details on Options
Note: these are AI generated and gently edited, for more information, see individual tool documentation
mmseqs_prot_cov
The mmseqs_prot_cov option sets the minimum protein coverage threshold for sequence comparisons. It is defined as a decimal number between 0 and 1, with a default value of 0.3. This threshold determines the minimum fraction of a protein sequence that must align with another sequence to be considered a significant match. A higher value results in more stringent criteria for sequence similarity.
mmseqs_prot_id
The mmseqs_prot_id option specifies the minimum protein identity threshold for sequence comparisons. It is defined as a decimal number between 0 and 1, with a default value of 0.99. This threshold sets the minimum sequence similarity required for two proteins to be considered related. A higher value indicates a stricter requirement for sequence identity.
mmseqs_prot_cov_mode
The mmseqs_prot_cov_mode option determines the mode for calculating protein coverage. It is an integer value, with a default setting of 1. Different modes may influence how protein coverage is computed, affecting the interpretation of sequence matches.
mmseqs_contig_cov
The mmseqs_contig_cov option sets the minimum contig coverage threshold for sequence comparisons. Contigs are typically longer sequences assembled from shorter reads. This parameter, with a default value of 0.6, determines the fraction of a contig that must align with another sequence to be considered a significant match.
mmseqs_contig_id
The mmseqs_contig_id option specifies the minimum contig identity threshold for sequence comparisons. Contig identity is similar to protein identity but applies to contig sequences. The default value is 0.99, and it determines the minimum sequence similarity required for two contigs to be considered related.
mmseqs_contig_cov_mode
The mmseqs_contig_cov_mode option, similar to mmseqs_prot_cov_mode, defines the mode for calculating contig coverage. It is an integer value, with a default setting of 1, which influences how contig coverage is calculated and impacts the interpretation of sequence matches.
max_overlap
The max_overlap option specifies the maximum allowable overlap between two contigs. It is expressed as a decimal number, with a default value of 0.5. This parameter is important for avoiding redundancy by excluding highly overlapping sequences.
min_orfs
The min_orfs option sets the minimum number of open reading frames (ORFs) required in a sequence. ORFs are segments of a DNA or protein sequence that have the potential to be translated into functional proteins. The default value is 1, meaning that a sequence must contain at least one potential ORF.